

PROGRAMMED cell death protein 1 (PD-1), a member of B7 family, is a transmembrane protein mainly expressed on T cells and tumor-infiltrating lymphocytes (TILs). Programmed death ligand 1 (PD-L1), shown to be a survivor factor, is a transmembrane protein intensively distributed in tumor cells, and its level is increased by pro-inflammation and administering interferon-γ (IFN-γ) during tumor treatment. The binding of PD-1 and PD-L1 maintains self-immune tolerance, induces T cell apoptosis, and contributes to tumor metastasis. ITSM and ITIM at the C- and N-terminals of PD-1, respectively, act as docking sites for both SHP-2 and SHP-1, through which PD-1 recruits tyrosine phosphatase, thus impeding T cell immune signaling and inducing immune suppression under the favor of PKCθ.1 PD-1 blocks the phosphorylation of Erk, Syk, and PLC, thereby inhibiting cytotoxic lymphocyte (CTL) activity and B cell function in peripheral blood.
The PD-1/PD-L1 axis is an important immune checkpoint in T cell exhaustion and interleukin-2 (IL-2) downregulation. PD-1/PD-L1 regulation is based on complex signaling networks, such as PI3K/Akt/mTOR and MAPK. By inhibiting ZAP70 recruitment, lck activation, and CD3ζ phosphorylation, PD-1/PD-L1 blocks PI3K/Akt activation and induces Gads/SOS/VAV expression, consequently leading to immune suppression.1 SHP2 participates in the MAPK network, which subsequently induces PD-1 expression and upregulates PD-L1 level. Furthermore, MEK inhibitors efficiently synergize with BRAF inhibitors during PD-1 inhibition. Besides, PD-1/PD-L1 level is also associated with gene expression modification, both epigenetically and post-translationally. Tumor cells remarkably induce PD-L1 expression and reduce CpG islands of immune checkpoint PD-L1. PD-1/PD-L1 high expression mimics endogenous checkpoints and downregulates E-cadherin, which facilitates tumor escape and cell proliferation.2 In contrast, CD80-Fc/PD-L1 overcomes immune suppression, and PD-1 binding to immunoglobins not only increases IFN-γ and IL-12 production, but also reverses PD-1/PD-L1-induced IL-5 and IL-13 reduction.3 In summary, the PD-1/PD-L1 axis can be treated as a target for tumor immunotherapy, and many crucial components of this axis, such as CDK5 and BRD4, could become promising PD-1/PD-L1 inhibitors in the future.
PD-1 and Tim-3 are overly expressed in tumor cells, and PD-1+Tim-3+ is associated with poor survival and shorter time to transformation, indicating rapid progression, refractoriness to treatment, and an overall dismal prognosis.4 In addition, a solid tumor with PD-L1 expression demonstrated a decrease in tumor immune surveillance. PD-L1 blockade inhibits the shift of Tregs toward Th17 cells and inhibits alloreactive T cell apoptosis. Actually, PD-1/PD-L1 is specifically regarded as a biomarker for tumor diagnosis and is a poor prognostic indicator of solid tumors.
Currently, PD-1/PD-L1 inhibitors, comprising monoclonal antibodies and small-molecule inhibitors, have been successfully applied in clinical trials to arrest tumor progression and prevent immune escape, with high stability and low adverse effects. This review aimed to summarize the main regulatory mechanisms of PD-1/PD-L1, current clinical applications of PD-1/PD-L1 inhibitors, and combination therapy of PD-1/PD-L1 inhibitors, in order to discover potential targets for tumor treatment.
EPIGENETIC AND POST-TRANSLATIONAL REGULATION OF PD-L1
PD-L1 level is closely related to epigenetic modification, which influences tumor-associated antigens expression and the related gene copy number. Tumor cells exhibit remarkably induced PD-L1 expression and reduced CpG islands of immune checkpoint PD-L1, subsequently suppressing Th2 cytokine production and increasing Th1 function. CpG-DNA strongly induces PD-L1 expression via NF-κB signaling and reduces level of inducible costimulator ligand.5 DNA modification may potentially help to downregulate PD-L1 expression and reverse immune suppression via methyltransferase and histone deacetylase inhibitors. Decitabine, a methylation inhibitor, has been shown to promote T cell activation with cytolysis capability and to block persistent T cell exhaustion within successive T-cell generations.6 MicroRNAs, such as miR-570, miR-34a, and miR-200, bind to specific mRNAs to prevent PD-L1 translation, participate in PD-L1 expression regulation, and induce epithelial-mesenchymal transition to promote metastasis.7
At the post-translational level, matrix metalloproteinases (MMPs), especially MMP-13, which is involved in extracellular matrix (ECM) degradation and tumor metastasis, modulates PD-L1 expression by cleaving the binding site of PD-1. Tumor-associated fibroblasts prevent PD-1 ligand depletion via MMP activity, possibly reversing apoptotic signals of mitogen-activated CD3+ T cells, while interfering exhausted memory T cells depletion.8 Glycogen synthase kinase 3β (GSK3β) binds to PD-L1 to mediate its phosphorylation, subsequently leading to PD-L1 ubiquitination and degradation in proteasomes. Besides, 17β-estradiol-induced binding of membrane estrogen receptor α (ERα) to the p85α regulatory subunit of PI3K activates the PI3K/Akt signaling pathway and increases PD-L1 mRNA stability and expression level.9
SIGNAL NETWORK OF PD-1 REGULATION
PI3K/Akt/mTOR
PD-1 impedes CD28-mediated ITSM phosphorylation, and CTLA-4 terminates Akt induction via PP2A, both of which block PI3K/Akt/mTOR pathway and inhibit the expression of anti-apoptotic genes such as Bxl (Fig. 1).10 PI3K inhibitors diminish resistance to BRAF inhibitors and induce PD-1 downregulation, suggesting that PD-1 positively correlate with PI3K. Moreover, many PI3K antibodies have currently been applied in clinical trials to downregulate PD-1 level. A pan-PI3Kδ inhibitor reportedly inhibited PD-1 and BKM120 significantly to facilitate interferon-γ secretion, with CD4+ T cell proliferation.11 Additionally, after administering Buparlisib (PI3K inhibitor), progressive free survival (PFS) among 63 patients with squamous carcinoma evidently improved (NCT01820325).
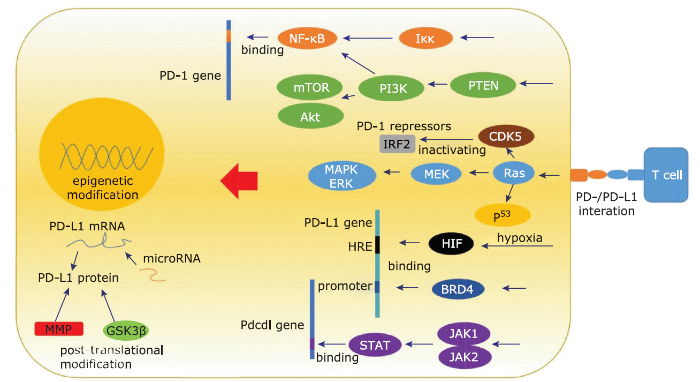
Figure 1.
The regulation network of PD-1/PD-L1.
PI3K/Akt/mTOR and MAPK signal pathway induces PD-1/PD-L1 expression, and transcriptional factors HIF, PTEN, p53, CDK5, BRD4, STAT etc modulate PD-1/PD-L1 expression. PD-1/PD-L1 level is also regulated via epigenetical and post-translational manner. GSK: glycogen synthase kinase; HRE: hypoxia response element; MMP: matrix metalloprotein.
PD-1-induced immune suppression downregulates Akt level, thereby contributing to T-cell receptor (TCR) normal functioning. Furthermore, an Akt inhibitor could accentuate PTEN loss with PD-1 upregulation. Clinically, an AKT inhibitor, MK-2206, induced apoptosis and estrogen deprivation, leading to an overall response rate of 15.4% among 13 patients with breast cancer.12 However, PD-1 inhibitors may upregulate Tim-3 level through Akt/PI3K, and Tim-3 cooperates with PD-1 in a synchronized manner, resembling a compensatory loop, in tumor models,13 indicating that Akt inhibitor monotherapy is not that effective; hence, a combination therapy involving multiple PD-1 agents should be adopted.
Mammalian target of rapamycin (mTOR) plays a crucial role in metabolism and contributes to anti-tumor effect. Raptor and rictor are the main components and regulators of the mTOR complex, and PD-1/PD-L1 binding may activate the Akt/mTOR signaling pathway in tumor cells to promote tumor progression. Furthermore, mTOR inhibitors synergize with PD-1 blockade during the clinical treatment of cancers; however, mTOR inhibitors may lead to side effects that enhance Tregs activity and subsequently reduce anti-tumor immunity. Temsirolimus (mTOR inhibitor) forms complexes with FKBP12 and inhibits its interaction with mTOR,14 and patients receiving temsirolimus showed partial response and prolonged disease stabilization.
MAPK
MAPK, characterized by its cascade reactions, is involved in tumor progression. SHP2 is a tyrosine phosphatase that participates in MAPK-induced PD-1 regulation and is likely to be a potential target. Furthermore, MEK inhibitors or knockdown of ERK1/2 efficiently synergizes with BRAF inhibitors in PD-1 inhibition and T cell infiltration, indicating that PD-1 is tightly related to the MAPK pathway, which activates epidermal growth factor receptor (EGFR) and upregulates PD-L1 level.7 Besides, combining IL-15 and a p38 MAPK inhibitor blocks redirection toward CD4+ Th17 cells, intensifies cytolysis effect, and downregulates PD-1/PD-L1, leading to a positive prognosis of ovarian tumors, indicating that MAPK associated tumor vaccine therapy is beneficial.15 Patients receiving a MEK inhibitor, selumetinib, showed well-tolerance, stable disease, and prolonged survival. However, innately resistant tumors display a transcriptional signature, with a high load of BRCA2 mutation in MAPK inhibitor therapy. The innate anti-PD-1 resistance and MAPK inhibition may establish a crosstalk in epithelial-mesenchymal transition, cell adhesion, and extracellular matrix (ECM) remodeling.16
TRANSCRIPTIONAL FACTORS OF PD-1/PD-L1 REGULATION
NF-κB
NF-κB, an important nuclear transcriptional factor and inflammatory mediator, is involved in stress and inflammation. NF-κB-related gene elements undergo degradation in tumor cells, making anti-tumor effects less efficient. Regarding NF-κB mediated PD-1 regulation, NF-κB binding sites exist in conserved region C and p65, which lay the foundation for PD-1 initiation in macrophages under the guidance of T-cell receptor (TCR) activation and Toll-like receptor (TLR) ligands. Moreover, NFAT blockade by cyclosporin inhibits PD-1 production, indicating that NFAT is a critical inducer of PD-1 expression.17 PD-1 has been shown to inactivate NF-κB and plays an inhibitory role in an SH2-independent manner, thereby promoting immune evasion and inhibiting the function of tumor-infiltrating dendritic cells. Further experiments using gefitinib, an EGFR-TKI inhibitor, or siRNA to knockdown p65, indicate that PD-L1 expression relies on NF-κB via IFN secretion.18 Regarding the clinical use of NF-κB inhibitors, caffeic acid phenethyl ester with tetrodotoxin could remarkably reduce lateral motility and invasion of tumors by inhibiting voltage-gated sodium channels activity.19 Stimulator of interferon genes (STING) activates NF-κB and STAT pathways to facilitate proinflammation, and STING vaccine plus NF-κB blockade can improve tumor regression and induce a potent anti-tumor effect.
HIF
Hypoxia induced factor (HIF) is a heterodimer, which is constituted of α subunit and β subunit. HIF cooperates with CBP/P300 and binds to hypoxia response element (HRE) on PD-L1 promoter to initiate adaptation to hypoxia. PD-1 enhances HIF-1α level and inhibits ubiquitin mediated HIF-1 degradation to make HIF more stable. ERO1-α is the oxidase of endoplasmic reticulum and acts as an indicator of hypoxia. ERO1-α gives rise to PD-1 production and promotes oxidative protein folding.20 HIF induces PD-1 upregulation, which promotes resistance to cytolysis effect and contributes to T cell dysfunction. And PD-1 blockade delays tumor progression without interfering TILs function, which enhances PPAR-α level and fatty acid catabolism in hypoxic cells. PD-1 mediated angiogenesis with HIF collaboration manifests poor prognosis and HIF can be treated as direct target for PD-1 inhibition. HIF inhibitor (LW6) is benzimidazole analogue without severe side effects and reduces tumor lesion by 58.6% via Hsp90-Akt pathway.21 Applying nitric oxide (NO) signaling agonist glyceryl trinitrate also blocks HIF-1α accumulation and mitigates immune escape with reduced level of IL-10 and diminished resistance to cytolysis.22
CDK5
The CDK family generally regulate the cell cycle, and CDK5, a unique member of the family, binds to p35 or p39 to maintain activation of the central nervous system (CNS). CDK5 determines PARP activity and associates with STAT3 at the C-terminal to participate in DNA repair and replicative stress response, indicating its direct role in cell cycle checkpoint.23 K-Ras mutation-induced CDK5-p35 complex is overly amplified in pancreatic carcinoma, which is associated with angiogenesis and chemotherapy resistance.24 Conversely, knockdown of CDK5 via siRNA arrests cell cycle and enhances TP53 level. CDK5 deficiency contributes to persistent hyperphosphorylation of PD-1 transcriptional repressors (IRF2BP2 and IRF2), which depress PD-L1 level and enhance anti-tumor immunity of CD4+ T cells. In addition, c-myc-mediated mutant expression of EGFR and β-catenin triggers PD-L1 expression, with the absence of T cell signature; conversely, CDK5 disruption reduces PD-L1 level. CDK5 serves as a potential target in inhibiting the PD-1/PD-L1 axis and anti-tumor treatment. Dinaciclib is a novel inhibitor of CDK1/2/5/9, and after administrating dinaciclib, partial response rate in patients with relapsed multiple myeloma reached 11% (NCT01096342).
PTEN
PTEN is an important tumor-suppressor gene and acts as a negative regulator of the PI3K/Akt/mTOR pathway. Conventionally, PI3K/Akt activation in tumors is closely accompanied with PTEN loss, which contributes to a high PD-L1 expression level and constraints T cell infiltration. PI3K blockade (rapamycin), which decreases PD-L1 expression, indicates a tight link between PTEN and PD-L1. PTEN, phosphorylated by CK2 at the C-terminal regulatory domain, could be more stable; however, PD-1 recruits PTEN phosphatase and accelerates ubiquitin-dependent degradation of PTEN.25 Therefore, CK2 may be considered as a potential target of PD-1. PTEN loss is a novel player in PD-L1 regulation, and thus can be used as a biomarker for tumor diagnosis and as a treatment target to ameliorate PD-L1 expression from PTEN perspective. PTEN may also act as a potential candidate target for blocking the PD-L1 axis in gastric cancer using antibodies.
TP53/p53
The wild-type of p53 cooperates with p21 to arrest the cell cycle and is regulated by post-translational modifications, while the abnormal phosphorylation of p53 disrupts p53-MdM2 interaction, with p53 stabilization and accumulation. p53 is closely related to PD-L1 level in tumors, and PD-L1 expression is positively related to p53 aberrant expression in lung adenocarcinoma. IFN induces TP53, K-RAS, and epidermal growth factor receptor (EGFR)-vIII mutations to jointly upregulate PD-L1 level and alter the expression of genes involved in the cell cycle. p53 also modulates PD-L1 expression through miR-200 and suppresses the effect of PD-L1 in NK cell and T cell recognition. Besides, via miR-34, p53 modulates transforming growth factor (TGF-β), CCL22 (chemokine C-C motif ligand 22), DGKζ (diacylglycerol kinase ζ), and Tregs activity.26 Furthermore, after administering PD-1 inhibitors, the pathological status of patients showed a remarkable progress in limiting cancer metastasis.
BRD4
BRD4, a member of the BEF family, binds to an epigenetic reader and is involved in autophagy, inflammation, and lysosomal function. BRD4 localizes to P-TEFb complexes consisting of Cdk9 and cyclin, which play a central role in RNA polymerase Ⅱ promoter-proximal pause release and phosphorylation under the favor of acetylated histones.27 C-myc binds to CD47 (PD-1) promoter and PD-L1 to facilitate downstream molecules production. JQ1, a co-inhibitor of BRD4 and CHK1, mediates c-myc functional suppression and reduces PD-1/PD-L1 level. In contrast, JQ1 affects specific lineage genes, such as CD4+ T cell promoter and LAG3 expressed on TIL, which hinder BRD4 from recognizing histones, to suppress PD-1/PD-L1 expression.28 Apart from the aforementioned, BRD4, epigenetic readers, and super-enhancers have been confirmed to have a strong association with gene copy number and high level of single nucleotide polymorphisms (SNPs) in breast cancer. In summary, BRD4 is a significant factor in tumor progression and is a direct target for blocking PD-1/PD-L1 axis transcription. Due to the enriched BRD4 at the CD274 gene promoter, the active expression of BRD4 in tumors contributes to high levels of CD274 and IFN-γ induced PD-1.29 Besides, bromodomain and external domain (BET) inhibitors could suppress c-myc gene expression, leading to PD-1/PD-L1 and CCR4 expression blocking. Therefore, a combination therapy of BET bromodomain kinase inhibitors can potentially help to control PD-1/PD-L1 level and to treat tumors.
STAT
Pdcd1 gene locus contains STAT/NFAT binding sites. PD-1 expression is associated with IL-6 and IL-12 levels, which are induced by STAT3 and STAT4 respectively. IFN sensitive response element leads to upregulation of PD-1/PD-L1 expression through the effect of STAT1/2, which lays foundation for STAT inhibitor (Ruxolitinib) induced anti-tumor immune response. JAK2 inhibitor or Ruxolitinib can enhance adaptive inmmune response, and selectively inhibit STAT1 and STAT3.15, 30 PD-1 blocks localization and polarization of M1 type macrophage via reducing STAT1 molecule, however, PD-1 enhances M2 type macrophage activity through STAT6.31 Besides, IFN-γ produced by NK cell activates STAT1 and increases resistance of PD-L1 to NK cell lysis. This indicates JAK/STAT inhibitor may cooperate with PD-1/PD-L1 monoclonal antibody to prevent tumor cytolysis, which might cause serious immune disorder. In addition, phosphorylation of STAT pathway is associated with PD-L1 transcription, which is stimulated by chemotherapy agent like 5-flurouracil in dosage dependent manner.32
IMMUNOTHERAPY WITH PD-1/PD-L1 INHIBITOR IN CLINICAL TUMOR TREATMENT
In recent years, cancer researches on PD-1/PD-L1 and its related inhibitors have gradually become the new focuses, and most of inhibitors have been reported with clinical efficacy as well as potential adverse effects. Long-term survival rate only occurred in small scale of patients who received ipilimumab and the overall response rate is still concerned. Regarding immune suppressive microenvironment induced by PD-1/PD-L1, therefore we recommend combination use of PD-1/PD-L1 agents instead of monotherapy.
Pembrolizumab
Pembrolizumab, the first-line PD-1 inhibitor agent, is a highly selective and humanized monoclonal antibody, which binds to PD-1 receptor to prevent interaction of PD-1 with PD-L1, and stimulates anti-tumor immune response. Pembrolizumab has been administrated in melanoma treatment with a progression free survival rate of 47.3% in 6 months (NCT01866319). A total of 23 patients with colorectal cancer were selected for pembrolizumab trial in 2014. Overall response rate was 4%, 1 patient in this cohort experienced partial response, and 4 patients had stable diseases (NCT02054806). However, the adverse effects could not be ignored with rate of 70.9%, which manifest as fatigue, decreased appetite, and hypothyroidism. These adverse effects could be ameliorated via managing dosage with cautious consideration and administrating corticosteroids to reduce inflammation.
Nivolumab
Nivolumab is also a humanized IgG4 monoclonal antibody of PD-1. It is deemed safe in dosage use and observed with median overall survival for 12.2 months among 292 patients in non-small cell lung cancer (NSCLC) treatment.33 Compared to other anti-PD1 agents like docetaxel, nivolumab administration prolongs overall survival rate and shows less severity including fatigue, nausea, and asthenia, which could be possibly reversed by immune modulators like glucocoticoids.
Atezolizumab
Atezolizumab is anti-PD-L1 antibody, and has been clinically applied in renal carcinoma and NSCLC treatment, which presents superiority to docetaxel concerning death rate and adverse effects. Atezolizumab obviously improves IFN-γ expression in TILs and increases overall survival rate among patients with PD-L1 positive tumor. In atezolizumab group, overall survival of 63 patients with renal clear cell carcinoma reached 28.9 months, objective response rate reached 15%, and progression free survival lasted for 5.6 months (NCT01375842).
Ipilimumab
Ipilimumab is a kind of anti-CTLA4 antibody, has become a standard practice in treatment of metastatic melanoma, prostate carcinoma, and small cell lung cancer, which improves overall survival. In phase 3 of anti-melanoma clinical trial, ipilimumab monotherapy plus dacarbazine improved the overall survival, however, clinical response rate was still low and only small scale of patients achieved the long-term survival rates.34 Ipilimumab commonly coordinates with nivolumab in clinical trials and has achieved success. In PD-L1 negative patients, the combination therapy of nivolumab plus ipilimumab demonstrates superiority in higher progressive free survival to monotherapy, while PD-L1 positive patients would not be sensitive to this. Complete response rate reached 22%, objective response attained 61% in the combination group, and no complete responses were demonstrated in patients of the ipilimumab monotherapy group (NCT01927419).
Combination therapy
It has been proved combining several blockades simultaneously can achieve higher response rate and mitigate tumor escape, therefore combination therapy is gaining prominence in tumor treatment. (1) Combination therapy of first-line and second-line PD-1/PD-L1 inhibitors simultaneously can increase tolerance of patients and decrease toxicity. Pembrolizumab plus ipilimumab combination therapy has been practiced among 153 patients, PFS was 69% in 1 year and overall survival was 89% (NCT02089685). (2) Combining more than two checkpoint monoclonal antibodies, like ipilimumab/nivolumab, would provide a novel therapeutic approach to increase efficacy and decrease toxicity. The overall survival rate reached 58% in nivolumab-ipilimumab combination cohort, as compared with 34% in the ipilimumab mono-group and 52% in the nivolumab mono-group (NCT01844505). (3) PD-1/PD-L1 monoclonal antibody in combination with tumor vaccine also offers a fresh outlook for melanoma treatment. The median relapse-free survival among patients who received both nivolumab and tumor vaccine was 47.1 months, and overall survival rate reached more than 80%.35 (4) There also emerges a trend that redirected T cell (CAR-T) plus PD-1 blockade would favor tumor treatment. Recently, anti-GD2 specific CAR plus pembrolizumab therapy has been observed with long persistence and limited immune escape (NCT01822652). (5) Indoleamine 2, 3-dioxygenase 1 (IDO1) involves in tryptophan catabolism and facilitates immunosuppressive microenvironment. Erk/p38/MAPK signaling in dendritic cells is associated with PD-L1 reduction and IDO inactivation. IDO1 overexpression accompanies with immune escape, and IDO1 inhibition plays a crucial role in anti-tumor surveillance and prognosis. At present, PD-1/PD-L1 and IDO1 double inhibition indicates a higher benefit and a potential trend for tumor treatment. Among 19 patients who received combination therapy of IDO inhibitor (epacadostat) plus pembrolizumab, 4 were observed with complete responses, 7 with partial responses, and 3 with stable diseases (NCT02178722). (6)Apart from specific PD-1/PD-L1 monoclonal antibodies (mAbs), small molecule inhibitors of PD-1/PD-L1 undergo slow but emerging preclinical development, which incorporate multiple disciplines. Via binding to PD-1/PD-L1, small molecule inhibitors present variable structures and better tumor infiltration capability. Based on data of compounds like BMS-202 applied in clinical treatment, small molecule inhibitors have been structurally and biochemically designed to induce PD-L1 dimerization, which subsequently impede PD-1 pathway.36 Additionally, small molecule GSK-3α/β inactivation by siRNA may increase NFAT in nucleolus, which contains Pdcd1 transcription regulator and consequently inhibits PD-1 expression without influencing CTL killing effect.37 Besides, BET plays a crucial role in PD-1/PD-L1 expression, thus may serve as a novel clinical target for PD-1/PD-L1+ tumor treatment. The BET inhibitor (FT-1101) has been testing its clinical efficacy among patients with relapsed/refractory myeloid leukemia at present (NCT02543879). (7) Modification of current monoclonal antibodies would be plausible for PD-1/PD-L1+ tumor treatment. MPDL-3280A is an engineered humanized anti-PD-L1 IgG1 mAb and the function of IgG1 Fc domain completely abolishes the antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Patients with advanced cutaneous melanoma who received MPDL-3280A had a response rate of 29%. In addition, BMS-936559 is a fully humanized anti-PD-L1 IgG4 mAb, which inhibits PD-1 and CD80/B7 binding, and 16% patients with melanoma had objective responses.38
To sum up, PD-1/PD-L1 serves as an important immune checkpoint and is regulated via PI3K/Akt/mTOR and MAPK signal network. Transcriptional factors including NF-κB, HIF, PTEN, p53, CDK5, BRD4, STAT are critical factors influencing PD-1/PD-L1 expression. Besides, epigenetic and post-translational modification also participates in PD-1/PD-L1 regulation. All these could serve as potential targets, some of which have made clinical progress. One thing must be noted that seeking for managements of toxicity and adverse effects is imperative. Recently, PD-1/PD-L1 mAbs applied in treatment showed high efficacy and combination therapy would provide bright future for PD-1/PD-L1 immunotherapy.
Conflicts of interest statement
The authors declare that they have no competing interests.
参考文献
PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3 signalosome and downstream signaling to PKC theta
Prognostic significance of PD-L1 expression on tumor cells and tumor-infiltrating mononuclear cells in upper tract urothelial carcinoma
The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy
Pattern of CD14+ follicular dendritic cells and PD1+ T cells independently predicts time to transformation in follicular lymphoma
Cytosine-phosphate-guanosine-DNA induces CD274 expression in human B cells and suppresses T helper type 2 cytokine production in pollen antigen-stimulated CD4-positive cells
Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade

Immune checkpoint blockades, such as inhibitors against programmed death 1 (PD-1) and its ligand (PD-L1), have received extensive attention in the past decade because of their dramatic clinical outcomes in advanced malignancies. However, both primary and acquired resistance becomes one of the major obstacles, which greatly limits the long-lasting effects and wide application of PD-1/PD-L1 blockade therapy. PD-1/PD-L1 both regulates and is regulated by cellular signaling pathways and epigenetic modification, thus inhibiting the proliferation and effector function of T and B cells. The lack of tumor antigens and effective antigen presentation, aberrant activation of oncogenic pathways, mutations in IFN- signaling, immunosuppressive tumor microenvironment such as regulatory T cells, myeloid-derived suppressor cells, M2 macrophages, and immunoinhibitory cytokines can lead to resistance to PD-1/PD-L1 blockade. In this review, we describe PD-1 related signaling pathways, essential factors contributing to the resistance of PD-1 blockade, and discuss strategies to increase the efficacy of immunotherapy. Furthermore, we discuss the possibility of combined epigenetic therapy with PD-1 blockade as a potential promising approach for cancer treatment.
Regulation of PD-L1: a novel role of pro-survival signalling in cancer
A novel regulation of PD-1 ligands on mesenchymal stromal cells through MMP-mediated proteolytic cleavage
Posttranscriptional control of PD-L1 expression by 17β-estradiol via PI3K/Akt signaling pathway in ERα-positive cancer cell lines
Targeting the programmed death-1 pathway in lymphoid neoplasms
Programmed death-1 (PD-1) is a co-inhibitory molecule and is seen in CD4+ and CD8+ T cells. Upon binding to its ligands, programmed death ligand-1 (PD-L1) and -2 (PD-L2), PD-1 negatively regulates interleukin 2 (IL-2) production and T cell proliferation. Activated effector T-cells, which kill cancer cells, can be affected by PD-1 signaling in some lymphoid neoplasm that express PD-L1 or PD-L2. PD-L1 expression in tumor cells can be induced by extrinsic signal (i.e. interferon gamma) or intrinsic signals, such as genetic aberrations involving 9p24.1, latent Epstein arr virus infection, PD-L1 3 - untranslated region disruptions, and activated Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Anti-PD-1 therapy improves the overall response rate to treatment in patients with lymphoid neoplasms, particularly relapsed/refractory classical Hodgkin lymphoma. Inspired by their success in treating patients with classical Hodgkin lymphoma, medical practitioners have expanded PD-1 therapy, given as a single therapy or in combination with other drugs, to patients with other types of lymphoma. In this review, current clinical trials with anti-PD-1 or anti-PD-L1 drugs are summarized. The results of numerous clinical trials will broaden our understanding of PD-1 pathway and shall expand the list of patients who will get benefit from these agents including those who suffer from lymphoid neoplasms.
PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses
A phase I study of the AKT inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor-positive metastatic breast cancer
Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer
The prevalence and impact of hyperglycemia and hyperlipidemia in patients with advanced cancer receiving combination treatment with the mammalian target of rapamycin inhibitor temsirolimus and insulin growth factor-receptor antibody cixutumumab
Modulation of p38 MAPK signaling enhances dendritic cell activation of human CD4+ Th17 responses to ovarian tumor antigen
Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma
High mutational loads are associated with improved survival in melanoma patients but are not predictive of response to anti-PD-1 therapy, suggesting that other genomic and non-genomic features also contribute to response patterns on PD-1 checkpoint blockade therapy.
NF-kappaB regulates PD-1 expression in macrophages
ABSTRACT
EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB
Non-small-cell lung cancer (NSCLC) is a severe disease threatening human health. Targeted therapy of epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) has obtained potent efficacy in the treatment of NSCLC patients. However, the effects of EGFR-TKIs on tumor immune microenvironment are unclear. In this study, we show that NSCLCs with EGFR mutation express higher programmed cell death ligand 1 (PD-L1) than NSCLCs with wild type EGFR. The EGFR activation is also associated with high expression of PD-L1. The EGFR-TKI gefitinib can reduce PD-L1 expression, via inhibiting NF- B, in EGFR mutant NSCLC invitro and invivo . These findings elucidate a novel anti-tumor mechanism of EGFR-TKI and provide the possibility of combined strategy of targeted therapy and immunotherapy for EGFR mutant NSCLC patients.
Caffeic acid phenethyl ester: inhibition of metastatic cell behaviours via voltage-gated sodium channel in human breast cancer in vitro
Cancer-associated oxidoreductase ERO1-alpha promotes immune escape through up-regulation of PD-L1 in human breast cancer
Chemical biology approach for the development of hypoxia inducible factor (HIF) inhibitor LW6 as a potential anticancer agent
A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells
Abstract Immune escape is a fundamental trait of cancer in which mechanistic knowledge is incomplete. Here, we describe a novel mechanism by which hypoxia contributes to tumoral immune escape from cytotoxic T lymphocytes (CTL). Exposure of human or murine cancer cells to hypoxia for 24 hours led to upregulation of the immune inhibitory molecule programmed cell death ligand-1 (PD-L1; also known as B7-H1), in a manner dependent on the transcription factor hypoxia-inducible factor-102± (HIF-102±). In vivo studies also demonstrated cellular colocalization of HIF-102± and PD-L1 in tumors. Hypoxia-induced expression of PD-L1 in cancer cells increased their resistance to CTL-mediated lysis. Using glyceryl trinitrate (GTN), an agonist of nitric oxide (NO) signaling known to block HIF-102± accumulation in hypoxic cells, we prevented hypoxia-induced PD-L1 expression and diminished resistance to CTL-mediated lysis. Moreover, transdermal administration of GTN attenuated tumor growth in mice. We found that higher expression of PD-L1 induced in tumor cells by exposure to hypoxia led to increased apoptosis of cocultured CTLs and Jurkat leukemia T cells. This increase in apoptosis was prevented by blocking the interaction of PD-L1 with PD-1, the PD-L1 receptor on T cells, or by addition of GTN. Our findings point to a role for hypoxia/HIF-1 in driving immune escape from CTL, and they suggest a novel cancer immunotherapy to block PD-L1 expression in hypoxic-tumor cells by administering NO mimetics.
Cdk5 promotes DNA replication stress checkpoint activation through RPA-32 phosphorylation, and impacts on metastasis free survival in breast cancer patients
Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras
PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2
PDL1 regulation by p53 via miR-34
Although clinical studies have shown promise for targeting PD1/PDL1 signaling in non–small cell lung cancer (NSCLC), the regulation of PDL1 expression is poorly understood. Here, we show that PDL1 is regulated by p53 via miR-34. p53 wild-type and p53-deficient cell lines (p53–/–and p53+/+HCT116, p53-inducible H1299, and p53-knockdown H460) were used to determine if p53 regulates PDL1 via miR-34. PDL1 and miR-34a expression were analyzed in samples from patients with NSCLC and mutated p53 vs wild-type p53 tumors from The Cancer Genome Atlas for Lung Adenocarcinoma (TCGA LUAD). We confirmed that PDL1 is a direct target of miR-34 with western blotting and luciferase assays and used a p53R172HΔg/+K-rasLA1/+syngeneic mouse model (n = 12) to deliver miR-34a–loaded liposomes (MRX34) plus radiotherapy (XRT) and assessed PDL1 expression and tumor-infiltrating lymphocytes (TILs). A two-sidedttest was applied to compare the mean between different treatments. We found that p53 regulates PDL1 via miR-34, which directly binds to thePDL13’ untranslated region in models of NSCLC (fold-change luciferase activity to control group, mean for miR-34a = 0.50, SD = 0.2,P< .001; mean for miR-34b = 0.52, SD = 0.2,P= .006; and mean for miR-34c = 0.59, SD = 0.14, andP= .006). Therapeutic delivery of MRX34, currently the subject of a phase I clinical trial, promoted TILs (mean of CD8 expression percentage of control group = 22.5%, SD = 1.9%; mean of CD8 expression percentage of MRX34 = 30.1%, SD = 3.7%,P= .016, n = 4) and reduced CD8+PD1+cells in vivo (mean of CD8/PD1 expression percentage of control group = 40.2%, SD = 6.2%; mean of CD8/PD1 expression percentage of MRX34 = 20.3%, SD = 5.1%,P= .001, n = 4). Further, MRX34 plus XRT increased CD8+cell numbers more than either therapy alone (mean of CD8 expression percentage of MRX34 plus XRT to control group = 44.2%, SD = 8.7%,P= .004, n = 4). Finally, miR-34a delivery reduced the numbers of radiation-induced macrophages (mean of F4-80 expression percentage of control group = 52.4%, SD = 1.7%; mean of F4-80 expression percentage of MRX34 = 40.1%, SD = 3.5%,P= .008, n = 4) and T-regulatory cells. We identified a novel mechanism by which tumor immune evasion is regulated by p53/miR-34/PDL1 axis. Our results suggest that delivery of miRNAs with standard therapies, such as XRT, may represent a novel therapeutic approach for lung cancer.
Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release
Abstract Distal enhancers characterized by the H3K4me(1) mark play critical roles in developmental and transcriptional programs. However, potential roles of specific distal regulatory elements in regulating RNA polymerase II (Pol II) promoter-proximal pause release remain poorly investigated. Here, we report that a unique cohort of jumonji C-domain-containing protein 6 (JMJD6) and bromodomain-containing protein 4 (Brd4) cobound distal enhancers, termed anti-pause enhancers (A-PEs), regulate promoter-proximal pause release of a large subset of transcription units via long-range interactions. Brd4-dependent JMJD6 recruitment on A-PEs mediates erasure of H4R3me(2(s)), which is directly read by 7SK snRNA, and decapping/demethylation of 7SK snRNA, ensuring the dismissal of the 7SK snRNA/HEXIM inhibitory complex. The interactions of both JMJD6 and Brd4 with the P-TEFb complex permit its activation and pause release of regulated coding genes. The functions of JMJD6/ Brd4-associated dual histone and RNA demethylase activity on anti-pause enhancers have intriguing implications for these proteins in development, homeostasis, and disease. Copyright 2013 Elsevier Inc. All rights reserved.
PD-L1 is a therapeutic target of the bromodomain inhibitor JQ1 and, combined with HLA class I, a promising prognostic biomarker in neuroblastoma
BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression
Abstract Restoration of anti-tumor immunity by blocking PD-L1 signaling through the use of antibodies has proven to be beneficial in cancer therapy. Here, we show that02BET bromodomain inhibition suppresses PD-L1 expression and limits tumor progression in ovarian cancer. CD274 (encoding PD-L1) is a direct target of02BRD4-mediated gene transcription. In mouse models, treatment with the BET inhibitor JQ1 significantly reduced PD-L1 expression on tumor cells and tumor-associated dendritic cells and macrophages, which correlated with an increase in the activity of anti-tumor cytotoxic T02cells. The BET inhibitor limited tumor progression in a cytotoxic T-cell-dependent manner. Together, these data demonstrate a small-molecule approach to block PD-L1 signaling. Given the fact that BET inhibitors have been proven to be safe with manageable reversible toxicity in clinical trials, our findings indicate that pharmacological BET inhibitors represent a treatment strategy for targeting PD-L1 expression. Copyright 08 2016 The Author(s). Published by Elsevier Inc. All rights reserved.
JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer
Programmed death 1 deficiency induces the polarization of macrophages/microglia to the M1 phenotype after spinal cord injury in mice
The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines
Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer
Ipilimumab plus dacarbazine for previously untreated metastatic melanoma
Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma
Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2
Cancer cells can avoid and suppress immune responses through activation of inhibitory immune checkpoint proteins, such as PD-1, PD-L1, and CTLA-4. Blocking the activities of these proteins with monoclonal antibodies, and thus restoring T cell function, has delivered breakthrough therapies against cancer. In this review, we describe the latest work on structural characterization of the checkpoint proteins, their interactions with cognate ligands and with therapeutic antibodies. Structures of the extracellular portions of these proteins reveal that they all have a similar modular structure, composed of small domains similar in topology to the domains found in antibodies. Structural basis for blocking the PD-1/PD-L1 interaction by small molecules is illustrated with the compound BMS-202 that binds to and induces dimerization of PD-L1.
Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co-receptor PD-1 to enhance CD8(+) cytolytic T cell responses
The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma
Abstract PURPOSE: Blocking the interaction between the programmed cell death (PD)-1 protein and one of its ligands, PD-L1, has been reported to have impressive antitumor responses. Therapeutics targeting this pathway are currently in clinical trials. Pembrolizumab and nivolumab are the first of this anti-PD-1 pathway family of checkpoint inhibitors to gain accelerated approval from the US Food and Drug Administration (FDA) for the treatment of ipilimumab-refractory melanoma. Nivolumab has been associated with improved overall survival compared with dacarbazine in patients with previously untreated wild-type serine/threonine-protein kinase B-raf proto-oncogene BRAF melanoma. Although the most mature data are in the treatment of melanoma, the FDA has granted approval of nivolumab for squamous cell lung cancer and the breakthrough therapy designation to immune- checkpoint inhibitors for use in other cancers: nivolumab, an anti-PD-1 monoclonal antibody, for Hodgkin lymphoma, and MPDL-3280A, an anti-PD-L1 monoclonal antibody, for bladder cancer and non-small cell lung cancer. Here we review the literature on PD-1 and PD-L1 blockade and focus on the reported clinical studies that have included patients with melanoma. METHODS: PubMed was searched to identify relevant clinical studies of PD-1/PD-L1-targeted therapies in melanoma. A review of data from the current trials on clinicaltrial.gov was incorporated, as well as data presented in abstracts at the 2014 annual meeting of the American Society of Clinical Oncology, given the limited number of published clinical trials on this topic. FINDINGS: The anti-PD-1 and anti-PD-L1 agents have been reported to have impressive antitumor effects in several malignancies, including melanoma. The greatest clinical activity in unselected patients has been seen in melanoma. Tumor expression of PD-L1 is a suggestive, but inadequate, biomarker predictive of response to immune-checkpoint blockade. However, tumors expressing little or no PD-L1 are less likely to respond to PD-1 pathway blockade. Combination checkpoint blockade with PD-1 plus cytotoxic T-lymphocyte antigen (CTLA)-4 blockade appears to improve response rates in patients who are less likely to respond to single-checkpoint blockade. Toxicity with PD-1 blocking agents is less than the toxicity with previous immunotherapies (eg, interleukin 2, CTLA-4 blockade). Certain adverse events can be severe and potentially life threatening, but most can be prevented or reversed with close monitoring and appropriate management. IMPLICATIONS: This family of immune-checkpoint inhibitors benefits not only patients with metastatic melanoma but also those with historically less responsive tumor types. Although a subset of patients responds to single-agent blockade, the initial trial of checkpoint-inhibitor combinations has reported a potential to improve response rates. Combination therapies appear to be a means of increasing response rates, albeit with increased immune-related adverse events. As these treatments become available to patients, education regarding the recognition and management of immune-related effects of immune-checkpoint blockade will be essential for maximizing clinical benefit. Copyright 脗漏 2015 Elsevier HS Journals, Inc. All rights reserved.

{kind=link}
{kind=link}